-
Excited-state symmetry breaking in quadrupolar pull-push-pull molecules: dicyanovinyl vs. cyanophenyl acceptors
P. Verma, M. Tasior, P. Roy, S.R. Meech, D.T. Gryko and E. Vauthey
Physical Chemistry Chemical Physics, 25 (2023), p22689-22699


DOI:10.1039/D3CP02810K | unige:171173 | Abstract | Article HTML | Article PDF | Supporting Info
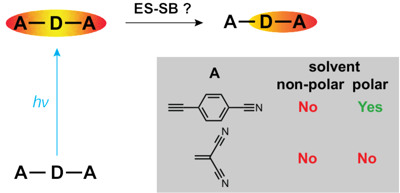
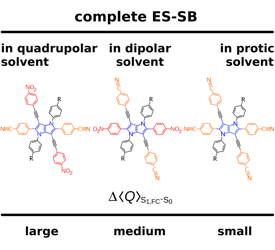
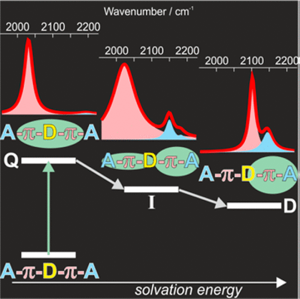
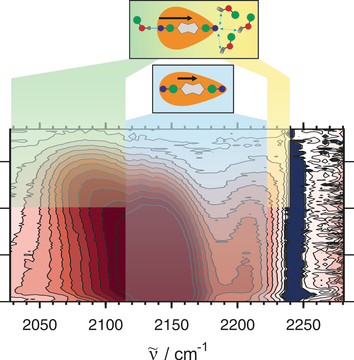
A significant number of quadrupolar dyes behave as their dipolar analogues when photoexcited in polar environments. This is due to the occurrence of excited-state symmetry breaking (ES-SB), upon which the electronic excitation, initially distributed over the whole molecule, localises preferentially on one side. Here, we investigate the ES-SB properties of two Aâââ‰â¬ÅDâââ‰â¬ÅA dyes, consisting of a pyrrolo-pyrrole donor (D) and either cyanophenyl or dicyanovinyl acceptors (A). For this, we use time-resolved vibrational spectroscopy, comparing IR absorption and femtosecond stimulated Raman spectroscopies. Although dicyanovinyl is a stronger electron-withdrawing group, ES-SB is not observed with the dicyanovinyl-based dye even in highly polar media, whereas it already takes place in weakly polar solvents with dyes containing cyanophenyl accepting groups. This difference is attributed to the large electronic coupling between the Dâââ‰â¬ÅA branches in the former dye, whose loss upon symmetry breaking cannot be counterbalanced by a gain in solvation energy. Comparison with analogues of the cyanophenyl-based dye containing different spacers reveals that interbranch coupling does not so much depend on the distance between the Dâââ‰â¬ÅA subunits than on the nature of the spacer. We show that transient Raman spectra probe different modes of these centrosymmetric molecules but are consistent with the transient IR data. However, lifetime broadening of the Raman bands, probably due to the resonance enhancement, may limit the application of this technique for monitoring ES-SB.